



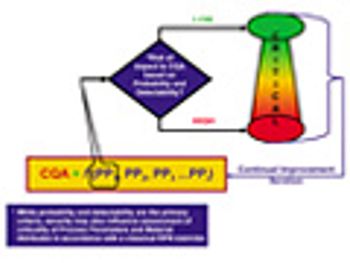
The authors provide an overview of the new ISPE Guide Series on Product Quality Lifecycle Implementation and how the guides can be used in a complementary way with existing guidance from FDA and the International Conference on Harmonization.
The evolution of therapeutic modalities drives the adoption of single-use technologies.

Despite its understandable hesitancy, the pharma industry is facing a need for more widespread adoption of cloud-based solutions.

Given the criticality of fill/finish processes, it is clear that automation is the next technological step.

The authors provide an overview of the new ISPE Guide Series on Product Quality Lifecycle Implementation and how the guides can be used in a complementary way with existing guidance from FDA and the International Conference on Harmonization.

A Q&A with Erik van den Berg, CEO of AM-Pharma, on recent industry trends.

Experts discuss the best practices for developing a QbD-based lyophilization process.

Soaring opioid use creates challenges for new drug development and supply-chain control.

China's drug-distribution network has been a mess for years, but government reforms and industry focus are unveiling new opportunities for market order and growth.

The contract provider needs to know as much as the NDA holder.

Pharmaceutical Technology Europe
Greece’s economic crisis has battered the country’s healthcare system, resulting in medicine shortages, market withdrawals and falling profits for the pharma industry.

Pharmaceutical Technology Europe
There are no two completely identical freeze dryer units in operation anywhere.

Pharmaceutical Technology Europe
Lyophilisation is often necessary for pharmaceutical products to improve stability or shelf-life. However, the process can present difficulties, particularly when scaling up from the laboratory to commercial production. We bring experts together to discuss best practices for developing a lyophilisation process, including quality by design (QbD) and design space.
Equipment and Processing Report
Modular factory concepts, which take flexible manufacturing to the next level, are beginning to take hold in the biopharmaceutical industry.

Equipment and Processing Report
PharmTech's monthly newsletter, Equipment and Processing Report, reviews the Editor's Picks for the March 2012 edition from EMD Millipore, ARTMiS, and Powder Systems.
Equipment and Processing Report
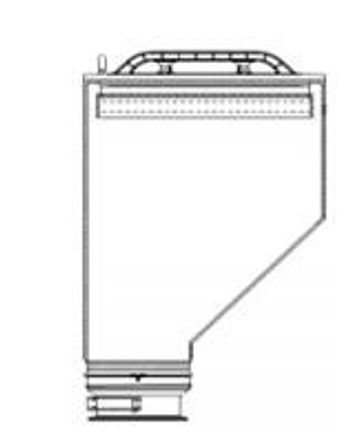
Powder-container design affects discharge rate, cleanability, and powder flow. Flow problems, such as ratholing, arching, and erratic flow, can be eliminated with appropriate container geometry.
Equipment and Processing Report
In our tablet coating process, we are losing up to 15% of our coating solution in each processing run. What can we do to prevent this problem in order to reduce waste and increase our cost efficiency?
“One of the most daunting challenges facing pharmaceutical companies is securing the long and complex supply chains that are typical in today’s global industry.
On Mar. 7, 2012, GE Healthcare announced an agreement to acquire Xcellerex, a supplier of manufacturing technologies for the biopharmaceutical industry, for an undisclosed amount.
GlaxoSmithKline and Daiichi Sankyo have formed a joint venture that they claim will create the biggest vaccines company in Japan. The joint venture will seek to improve access to vaccinations in the Asian nation as well as introduce new vaccines.

The standardization of upstream and downstream bioprocessing is growing, but several kinks need to be ironed out.

In a world where product recalls can mean the end of a company, all batches must be perfect.

Brazil's generic-drug market is growing steadily.
US Pharmacopeia documents best supply-chain practices and seeks broad input on proposal.

In Part II of a three-part article, the authors examine impurities from chiral molecules, polymorphic contaminants, and genotoxic impurities.

Has the long-awaited guidance answered all of the industry's questions?

Social media use raises questions about applying old standards to new information technology.

New product reviews for March 2012.

The evolving bio/pharmaceutical business model poses risk for CMOs.