

FDA explains the rewards that may be associated with switching from batch to continuous manufacturing.
Process and Scaling
Latest News
Advertisement
Advertisement
FDA approved an update in the manufacturing of Prezista (darunavir) using a continuous manufacturing line at Janssen Supply Chain’s facility in Puerto Rico.

Transferring the manufacturing of a drug from one scale to another or between manufacturing sites presents both technical and business challenges.
Pharmaceutical Technology spoke with Bill Randolph, vice-president, Technical Services, Janssen Supply Chain, about some of the considerations for technology transfer of a continuous, solid-dosage manufacturing process and what he sees as the outlook for continuous manufacturing.
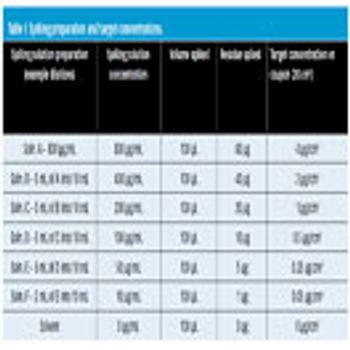
Visible residue limits have been shown to be a valuable tool in validated cleaning validation program.
A team of Bristol-Myers Squibb scientists will work in a new laboratory at the National Institute of Bioprocessing Research and Training facility in Dublin, Ireland.

The author discusses key process parameters and shows how a process optimization template can be used to improve this key operation.

The author discusses issues and best approaches for solid dosage form pharmaceuticals.

This article explores two commercial platforms, and touches on a new program underway in the UK.

Choosing the right facility size requires tailoring the design for current needs as well as anticipating the future.

The author discusses the collection and evaluation of data part of FDA’s definition of process validation.

DAVID LEAHY/GETTY IMAGESThe pharmaceutical industry has an important role to play in implementing solutions to global envi
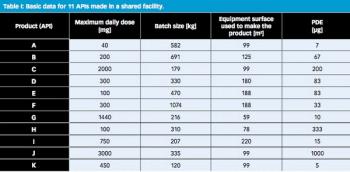
The 10-ppm criterion for the acceptable concentration of potential API in cleaning validation to minimize cross contamination into next product has been employed for many years. This article describes why the 10-ppm criterion, which was established based on analytical limitations and estimates of acceptability, is no longer necessary and why a risk-based approach should be universally adopted.
Quality by design, in-vitro release testing, and modern analytical methods are improving understanding and control of these complex formulations.
The FDA grants to the Rutgers-led C-SOPS research consortium will support the introduction of continuous manufacturing techniques for pharmaceuticals.
Siegfried Schmitt, principal consultant, PAREXEL, discusses how to assure compliance for automated systems.
Regulatory officials and industry scientists participated in a CMC Strategy Forum sponsored by CASSS in July 2015.
The new sizes follow the 2014 launch of the company’s fully disposable purifier.

An ISPE guidance document, four years in the making, brings risk-based thinking, statistics, and Lean Six Sigma to cleaning validation.
Barry Holtz, Principal, at Holtz Biopharma Consulting, and Klyo Collaborative, spoke with Pharmaceutical Technology about collaborative success strategies for biopharm companies.
The new column features Natrix’s signature macroporous hydrogel.
Presenters at IVT's Microbiology Week discussed best practices and recent guidance publications for microbial control in sterile and non-sterile pharmaceutical processes.
Talks will explore key trends and issues in the pharmaceutical space and address the “latest industry buzz.”

Optima Pharma is exhibiting a range of technologies from sterile-filling and freeze-drying solutions to isolators and restricted access barrier systems.

Hüttlin GranuLean offers a compact solution for high-quality granulation processes.
Advertisement
Advertisement
Trending on Pharmaceutical Technology
1
FDA Grants Priority Review For Pfizer’s Marstacimab for Hemophilia A or B
2
Drug Digest: Outsourcing Partnerships Fuel Global Biopharma Discovery and Scale-Up
3
PharmTech Weekly Roundup - February 6, 2026
4
Women in STEM: Early Phase Drug Development
5