
Medicago's new production facility will make plant-based vaccines and therapeutics.
Process and Scaling
Latest News
Advertisement
Advertisement
Economic benefits, equipment availability, research results, and FDA support are driving progress in continuous processing.
The VERISEQ nucleation technology offers a commercially viable technique for cryogenically generating a uniform dispersion of microscopic ice crystals (or ice-fog).
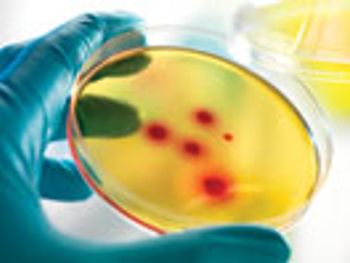
The author reports results of evaluations and concludes that a disinfectant composed of a low-concentration suspension of silver ions is completely sporicidal with only a one-minute contact time.
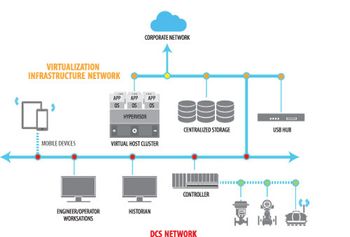
Virtualization has been mainstream in information technology (IT) for decades.
At the request of FDA, US Marshals seize unapproved prescription drugs from Florida distributor.
The document serves as guidance for the pharmaceutical industry in data integrity issues and complements existing EU GMP relating to APIs and dosage forms.
The European Medicines Agency releases guidelines for addressing and reporting risks associated with medication errors.

Industry awaits the final revision of USP General Chapter and the impact it will have on the evaluation of sterile product package integrity.

Quality-by-design tools improve efficiency in scale-up of pharmaceutical processes.
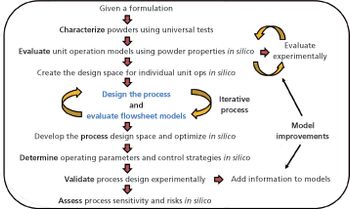
In-silico design facilitates process optimization and evaluation of process control strategies.

Process design experimental data and risk assessments are used to predict expected process performance and establish process performance qualification acceptance criteria.

The authors outline a validation approach for the manufacture of a “legacy product,” taking into consideration recent guidelines. The aim is to demonstrate the validity and capacity of the manufacturing process following a change in the manufacturer.
Multivariate data analysis (MVDA) is being used to effectively handle complex datasets generated by process analytical technology (PAT) in biopharmaceutical process development and manufacturing.
Use of modeling software can help improve process understanding, and can be used in open- or closed-loop control.
Real-time, noncontact imaging and spectroscopy techniques provide insight into pharmaceutical processes.
McNeil-PPC pleads guilty in connection with adulterated infants' and children's over-the-counter liquid medications.

The all-synthetic 3M Emphaze AEX Hybrid Purifier contains both an anion-exchange nonwoven media and a fine-particle, bioburden reduction membrane.
Working with biological matrices and understanding the intended use are crucial.
This article gives an overview of the concept and contents of the revised guidance and outlines how it has changed from the previous version.
New guidelines and best practices may lead to improved quality and reduced recalls due to visual defects.
Whether outsourcing or developing cell therapies in-house, success demands a focus on quality, cost of goods, and sustainability from the start.
Under an NIH contract, Paragon Bioservices will design a manufacturing process for recombinant human rhE-selectin protein.
Aurobindo Pharma USA issued a voluntary nationwide recall of Northstar Label Gabapentin Capsules, USP 300 mg, due to complaints of empty capsules.
The Baxter recall in the US of one lot of highly concentrated potassium chloride is due to a mislabeled overpouch.
Advertisement
Advertisement
Trending on Pharmaceutical Technology
1
FDA Grants Priority Review For Pfizer’s Marstacimab for Hemophilia A or B
2
Drug Digest: Outsourcing Partnerships Fuel Global Biopharma Discovery and Scale-Up
3
PharmTech Weekly Roundup - February 6, 2026
4
Women in STEM: Early Phase Drug Development
5