Lincoln Pharmaceuticals has received a patent, which is valid for 20 years, from the Government of India for its liquid diclofenac metered-dose rectal spray.
Regulatory Oversight and Compliance
Latest News
Advertisement
Advertisement
Pfizer has issued a voluntary recall of batches of its docetaxel injection 80 mg/8 mL due to impurity concerns.
A second lawsuit has been filed by Centrient Pharmaceuticals India in the High Court of Delhi in New Delhi against Dalas Biotech for patent infringement.
CHMP has recommended that Ervebo (rVSVΔG-ZEBOV-GP), a vaccine for active immunization against Ebola, be granted conditional marketing authorization in the EU.
CHMP has recommended that Baqsimi (glucagon), a non-injectable treatment for severe hypoglycemia, be granted marketing authorization in the EU.
The agency announced it has approved 1171 generic drugs in fiscal year 2019.
The guidance document discusses the preparation and submitting of drug master files as well as FDA’s review process.
The guidance discuses waivers, refunds, and reductions of user fees under sections 735 and 736 of the FD&C Act.
Over a two-year period, FDA inspections at company facilities found repeat problems with quality systems including lack of written procedures, inadequate investigation of out-of-specification conditions and corrective and preventive action, and insufficient cleaning and cleaning validation.
Sarah Hardison, head of Regulatory and Pharmacovigilance Solutions, Clarivate Analytics, will regulatory tackle trends in her presentation on the World of Pharma Podium during the second day of CPhI Worldwide.
A recently published European Medicines Agency (EMA) report has demonstrated that there are continued efforts being made across Europe to reduce the amount of antibiotics used in animals.
Emphasizing the need for senior management to take cGMP compliance seriously, the agency pointed to “improvisational” validation procedures, lack of compliance with written procedures, as well as inadequate process control, root cause analysis, and corrective and preventive action (CAPA) in its warning letter.
As the world awaits the outcome of Brexit, a presentation taking place at CPhI Worldwide in Frankfurt, Germany will delve deeper into the implications of each potential scenario on the pharma industry.
Innoveix Pharmaceuticals, Inc. is voluntarily recalling all sterile compounded drug products due to a lack of assurance of sterility.
The guidance describes an optional streamlined submission process for use of an investigational in vitro diagnostic in a clinical trial for oncology therapies.
AstraZeneca received approval from FDA for Fasenra Pen, a pre-fillled auto-injector pen that allows for self-administration of its asthma biologic therapy, Fasenra (benralizumab).
Glenmark Pharmaceuticals has been granted marketing approval from the Russian Ministry of Healthcare for its film-coated tablets, Montlezir, to treat seasonal and perennial allergic rhinitis.
The agency has approved Gilead Sciences’ Descovy (emtricitabine 200 mg and tenofovir alafenamide 25 mg) for reducing the risk of HIV-1 infection.
The tables can be used for new drug applications, biologics license applications, and supplements to these applications.
FDA and DEA warn four website operators to stop illegally selling opioids.
A new European Commission is likely, with the support of a new European Parliament, to give a higher profile to healthcare and pharmaceutical matters during its five years in office.
Falsified documents and manipulated test results prompt warnings and investigations.

In this article, experts discuss the fundamentals of dissolution testing and highlight the challenges that are surfacing as a result of the increasing numbers of poorly soluble molecules entering the development pipeline.
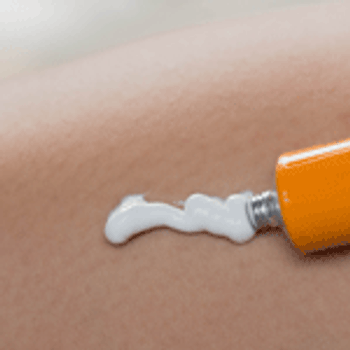
Changing regulatory guidance on generic topical products is expected to lead to an increase in the number of market approvals in the near future.
Data supporting the quality and safety of product must meet the ALCOA+ elements in order to avoid regulatory citations for data integrity issues, says Susan J. Schniepp, executive vice-president of post-approval pharma and distinguished fellow, Regulatory Compliance Associates.
Advertisement
Advertisement
Trending on Pharmaceutical Technology
1
Drug Digest: Outsourcing Partnerships Fuel Global Biopharma Discovery and Scale-Up
2
FDA Grants Priority Review For Pfizer’s Marstacimab for Hemophilia A or B
3
Shaping the Future of Microbial Development: From Gene to GMP
4
PharmTech Weekly Roundup - February 6, 2026
5