

As big pharmaceutical companies restructure and reevaluate the way they do business, contract research and manufacturing organizations could reap greater financial rewards.
Accurately targeted immunotherapies through reliable neoantigen recognition enable personalized medicine development.
Activation and expansion are essential for success in both autologous and allogeneic therapies.
Continuous manufacturing and a quality-by-design development approach are a natural fit.

As big pharmaceutical companies restructure and reevaluate the way they do business, contract research and manufacturing organizations could reap greater financial rewards.
Brussels (Feb. 13)-The European Generic Medicines Association raised concerns over what it terms ?the serious lack? of resources available to member states in the European Union (EU) to deal with the regulatory workload and bottleneck in new registrations caused by the new Decentralized Procedure (DCP) for approving drugs in Europe.
Enschede, Netherlands (Feb. 26)-Scientists from the University of Twente?s MESA+ Institute for Nanotechnology have designed a novel delivery system by combining synthetic iron-containing polymers with DNA macromolecules.
London (Feb. 22)-AstraZeneca unveiled further details of its plan to reduce staff at certain production facilities and announced a $120-million investment in a new process research and development (PR&D) facility in the United Kingdom.
Rockville, MD (Jan. 5)-The US Food and Drug Administration issued a Warning Letter to Bell-More Laboratories following the agency?s August 2006 inspection of the company?s Hampstead pharmaceutical facility.
Rockville, MD (Feb. 1)-The US Food and Drug Administration issued a revised warning letter to Actavis Totowa, LLC, citing ?significant deviations from the current Good Manufacturing Practice regulations.?

Large stability studies for drug products and devices with multiple strengths, packaging configurations and orientations can cost millions of dollars for the analytical testing alone.

Continuing an ambitious growth strategy, SAFC eyes CGMP manufacturing capacity in India for its custom-synthesis business and seeks to gain large-scale organics manufacturing capacity in China for raw-materials supply. And, its biosciences segment seeks opportunity in single-use disposable manufacturing.
InformexUSA, San Francisco (Feb. 14?16)-Several contract manufacturing organizations (CMOs) and technology providers used InformexUSA to announce plans to expand manufacturing capabilities, increase service offerings, and ramp up other investments.
InformexUSA, San Francisco (Feb. 12?Feb. 14)-Custom manufacturers of active pharmaceutical ingredients and intermediates are gathered at InformexUSA this week, with companies announcing investment plans and unveiling new technology
InformexUSA, San Francisco (Feb. 14)-NPIL Pharma unveiled a $100-million investment program in formulation development and manufacturing services.
InformexUSA, San Francisco (Feb. 14)-Almac Sciences plans to collaborate for cytotoxic fill-and-finish services with the Center for Pharmaceutical Science and Technology (CPST) at the University of Kentucky.
London, UK (Feb. 1)?Researchers at the University of London?s School of Pharmacy have chemically modified carbon nanotubes to enable them to enter human cancer cells.
St. Louis, MO, (Feb. 8)-SAFC, the custom manufacturing and fine chemicals unit of Sigma-Aldrich Corporation, is seeking to build a footprint in Asia, eying possible acquisitions in India and China.

As custom manufacturers gather for InformexUSA this month, strategies in asymmetric synthesis and catalysis prevail.

Sixty deals were completed in 2006, with a total value of $2 billion.

The same phenomena that create lightning and thunderstorms are around us every day, producing incredibly high voltages, which cause sparks and shocks. Static electricity is a mighty force. Each year excessive electrical charge build cause explosions in the grain industry.1 Look around any flammable storage area and you will see both grounding bars on the wall and cables, from the grounding bars connected to the drums of solvents. Take any material safety data sheet (MSDS) for a powder and look in section V; it highlights that any dry powder has the potential to attract and store a charge.
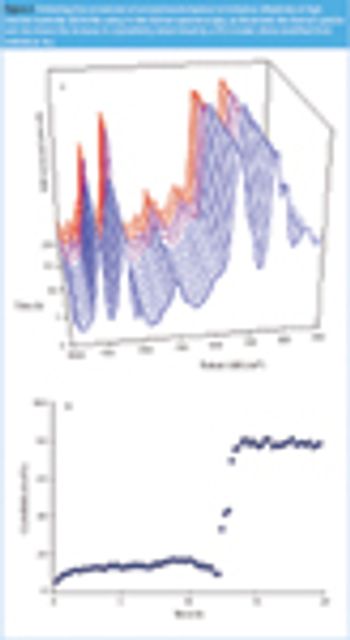
Raman spectroscopy has become a commonly used technique for physicochemical analysis that possesses many advantages over other analytical techniques. It is a very attractive characterization tool, not least because it enables measurements in water. However, very few examples of its application in an aqueous environment exist in literature. This paper provides some recent applications of Raman spectroscopy in pharmaceutical material and process characterization when water is present.
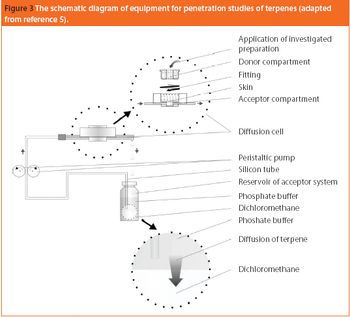
Terpenes are widely used in topical preparations. This paper focuses on the skin penetration of terpenes depending on their physicochemical properties and influence of the type of dermatological vehicle that was used. The structure of human skin and the skin penetration process is also briefly described.
Pfizer's restructuring plan provides yet another example of new supply-chain strategies by the pharmaceutical majors, which involve rationalization of manufacturing facilities and cost improvement. A review of these moves, an outlook for the pharmaceutical market in 2007, and analysis of US pharmaceutical production and trade.

Following the sales of its global branded pharmaceutical business, 3M stays the course in drug delivery as its Drug Delivery Systems Division advances technology in inhalation and transdermal drug delivery.

The recent struggles of Cardinal Health's and Patheon's contract dose manufacturing businesses offer a lesson on good business practices.
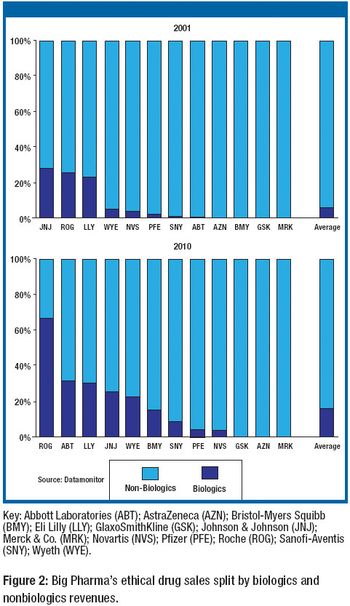
In 2007, the global pharmaceutical market is expected to grow moderately while biologics, generic drugs, and specialty-initiated drugs are projected to increase at double-digit rates. These trends for finished pharmaceuticals are reflected in the global market for active pharmaceutical ingredients (APIs), where the merchant generic API market is expected to see strong demand. On a production basis, India and China are forecast to raise their shares of the global generic API market against industry strongholds Italy and Spain. Meanwhile, the United States is expected to hold its its position as the leading producer of biotechnology-based APIs in an area traditionally dominated by captive production. And biogenerics or biosimilars gradually reshape the market.
During the past decade, the pharmaceutical industry has increased its use of information technology (IT) in research and development, production, and commercialization of pharmaceutical products. IT systems must be operated and maintained within a compliance-oriented framework to minimize risks; maximize safety and security, integrity, accuracy, reliability of information; and maintain product quality, For IT vendors and service providers, meeting requirements for qualification and validation calls for substantial investments in terms of creating capability, expertise, and resources. The author discusses the implications, challenges, and solutions in managing IT infrastructure qualification and validation in an FDA-regulated environment, particularly at vendor sites.
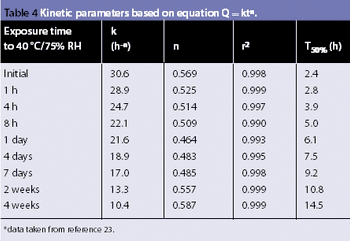
Water interacts with pharmaceutical solids at virtually all stages of manufacture, from synthesis of raw materials to the storage of the final dosage form. The interactions of water with powders is, therefore, a major factor in the formulation, processing and product performance of solid pharmaceutical dosage forms.