




The Mutual Recognition Agreement will allow FDA and EU inspectors to recognize each other’s work and avoid the duplication of drug inspections.
Process and Scaling
Latest News
Advertisement
Advertisement
Improved process analytical technology and new ways of thinking seek to enhance measurement and control for next-generation pharmaceutical manufacturing.
The agency sent a warning letter to Resonance Laboratories Pvt. Ltd. after an inspection found possible contamination problems.
Soft sensors are powerful tools that can be used along with spectroscopic instruments in on-line measurement.
GEA’s ConsiGma continuous tableting line combined with Siemens’ automation and Sipat data management systems enables continuous manufacturing.

As pharmaceutical quality metrics evolve, they will need to incorporate more of the principles of operational excellence, says consultant Prabir Basu.
The companies entered a manufacturing agreement for the future commercial production of Lenti-D and LentiGlobin product candidates.
Oxford Genetics received £1.61 million from Innovate UK to explore computational and synthetic biology approaches for optimized mammalian bioproduction.
FDA and BARDA awarded a contract to Continuus Pharmaceuticals to develop an end-to-end continuous manufacturing process for solid-dosage drugs.
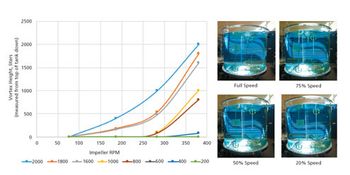
Quantitative and qualitative tools allow better understanding of mixing in a single-use bioprocessing system.

The half-cycle method for validating sterilization can have adverse effects on materials if used for steam sterilization.

The article proposes an integration of method validation, transfer, and verification process into the overall lifecycle management process.

A lifecycle approach can be used to develop GMP-compliant cleaning procedures for continuous manufacturing of solid-dosage pharmaceuticals.

Shear-sensitive ingredients in a tableting formulation experience different conditions in a batch tumble blender than they would in a continuous paddle blender.
Three-dimensional (3D) printing, which is a type of additive manufacturing (AM), enables fabrication of specialty drugs and medical devices, said Emil Ciurczak, Doramaxx Consulting and CPhI expert panel member, in the 2016 CPhI Annual Industry Report.

Flexible batch sizes for semi-continuous unit operations, such as tableting and encapsulation, can improve efficiency while maintaining quality.

A technology management process identifies and evaluates new technologies in biopharmaceutical manufacturing to aid business decisions.

Conventional limit-setting techniques are not health-based and can make risk assessment more difficult.

The success of a pharmaceutical manufacturing transfer from one facility to another requires detailed operational plans, attention to detail, and coordination between all parties. \
FDA issued a warning letter to the Worthing, UK facility for cross contamination and microbial contamination cGMP violations.
This new method uses inter-alpha inhibitors to promote attachment and long-term growth in stem cells.
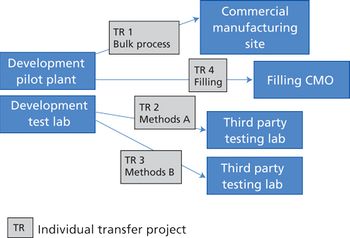
Integrating quality and compliance with technology transfer and careful project management are key in starting up a facility and launching a biologic drug.
FDA and bio/pharma companies get serious about continuous manufacturing to ensure product quality.

Industry experts discuss the benefits and challenges of using single-use systems in pharmaceutical manufacturing.

Experts discuss the key considerations in the development of an autoinjector.
Advertisement
Advertisement
Trending on Pharmaceutical Technology
1
Drug Digest: Outsourcing Partnerships Fuel Global Biopharma Discovery and Scale-Up
2
FDA Grants Priority Review For Pfizer’s Marstacimab for Hemophilia A or B
3
Shaping the Future of Microbial Development: From Gene to GMP
4
PharmTech Weekly Roundup - February 6, 2026
5