Rockville, MD (May 31)-The US Food and Drug Administration finalized guidances for seasonal and pandemic influenza vaccines, outlining the regulatory pathways for developing and approving these products.
Quality Systems
Latest News
Advertisement
Advertisement

Look ahead to keep from falling behind.

Product characterization and production challenges are key issues in developing a pathway for biosimilar therapies.

Perhaps Congress can help FDA meet its multiple oversight demands.

Signature authentication technology has evolved to become a stong tool for improving confidence in verification.

Boston, MA (May 8)-The global biotechnology industry showed several positive signs in 2006, including increases in overall revenues and financing, although the industry as a whole continues to operate at a loss, according to Ernst & Young's (New York, NY, www.ey.com) annual analysis of the biotechnology industry. Its report, "Beyond Borders 2007," was issued at the Biotechnology Industry Organization's (BIO, www.bio.org) annual conference and exhibition, which was held in Boston May 6–9.

The Active Pharmaceutical Ingredients Committee (APIC) - a sector group of Conseil European des Federations de l'Industrie Chimique (CEFIC) - first voiced the need for EU GMP API legislation in 1993 to help ensure the safety of medicines. In 2000, the International Conference on Harmonisation (ICH) finalized the harmonized API GMP Guideline Q7, which became legal in the US and Japan in 2001. The EU adopted a directive in March 2004 that includes the requirement for APIs in medicines for the EU market to comply with ICH/Q7A. Member States are transposing the directive into their national law: about half of them have completed this process, seven more are well on their way to completion, while seven others are still in earlier stages of adoption.
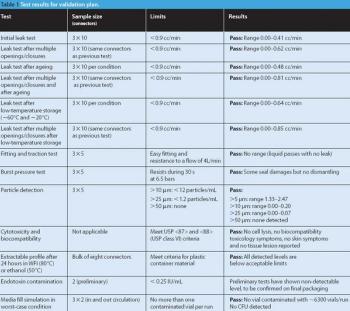
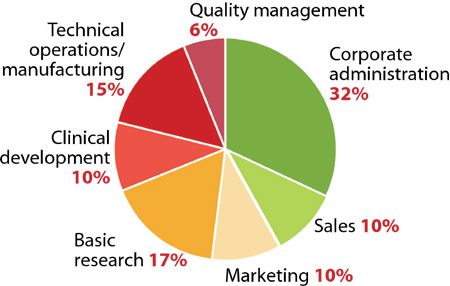
Sterile liquids are frequently transferred during the processing of sterile liquid drugs such as injectables or ophthalmic drops. Several types of transfer can be performed, each requiring a validated method to ensure the desired sterility-assurance levels are achieved.
It has been a long time coming, but stakeholders in the US are now seriously debating a route to market for cheaper copies of biopharmaceutical drugs. The European Agency for the Evaluation of Medicinal Products (EMEA) has led the way on this issue by publishing clear guidelines on what companies must do to get their versions of drugs such as erythropoietin (EPO), an advanced treatment for anæmia, and similar products approved.
Rockville, MD (May 25)-The US Food and Drug Administration issued a Form FDA 483, Inspectional Observations, to the Liverpool, United Kingdom, facility of MedImmune, Inc., citing deviations from current good manufacturing practices.
AAI, AstraZeneca, Biovail, West, more
Washington, DC (May 3)-The Biotechnology Industry Organization published a position paper stating that any legislation establishing a regulatory pathway for follow-on biologics should grant pioneering products 14 years of data exclusivity.
London (May 1)-The European Medicines Agency launched a database designed to facilitate the exchange of information about compliance with good manufacturing practices.
Washington, DC (April 30)-Less than two weeks after the Senate Health, Education, Labor, and Pensions Committee voted to reauthorize the Prescription Drug User Fee Act (PDUFA), the bill this week moved onto the Senate floor.
Rockville, MD (May 1)-The US Food and Drug Administration issued a guidance to alert pharmaceutical manufacturers, pharmacy compounders, repackers, and suppliers to the potential public health hazard of glycerin contaminated with diethylene glycol (DEG), a poison.
Breaking up is easy to do.

Fewer field offices and inspectors will increase reliance on manufacturers to ensure product and process quality.

Sanofi to Close Irish Plant, FDA Inspections and Warning Letters Continue to Decline, FDA Submits Final Proposals for PDUFA IV, and more.
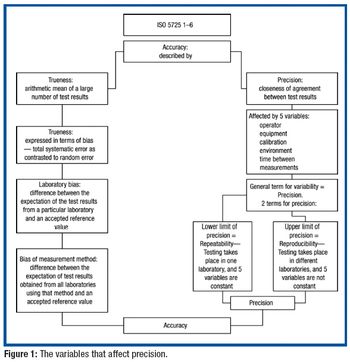
USP applies metrological principles to the dissolution procedure alone and in collaborative studies to understand and minimize potential sources of variability.
San Juan, PR (Apr. 25)-Regulatory and quality issues were prominent discussion topics at this year?s ExcipientFest conference and exhibition.
Interphex2007, New York, NY (Apr. 26)-Robustness studies for analytical methods are critical in being able to provide the assurance to the quality of an analytical method, a topic addressed in a conference session, "Performing Analytical Method Validation Robustness for Regulatory Compliance," at Interphex on Thursday.
Interphex2007 (Apr. 25)-As the pharmaceutical industry moves to a risk-based approach in manufacturing, analytics will play a critical role in not only meeting regulatory requirements but also in building needed collaboration between product development and manufacturing groups.
Interphex, New York, NY (Apr. 24)-Although the second revision of 21 CFR Part 11, the Electronic Records and Signatures Rule, has not been finalized, there is some progress being made. This was the topic of a presentation given by John English, manager of computer system validation for BE&K BioPharm at today?s Interphex Pharmaceutical Manufacturing Conference and Exhibition.
Lyon, France (Apr. 17)-Sanofi Pasteur, the vaccine division of the Sanofi-Aventis Group, announced that the US Food and Drug Administration has licensed its H5N1 vaccine, making it the first avian-influenza vaccine for humans in the United States.
Rockville, MD (Apr. 5)-The US Food and Drug Administration concluded its reinspection of Wyeth?s manufacturing facility in Guayama, Puerto Rico. Conditions at the Guayama facility prompted a Warning Letter in May 2006.
Advertisement
Advertisement
Trending on Pharmaceutical Technology
1
Fresenius Kabi and Phlow Corp. Enter First US End-to-End Manufacturing Collaboration
2
Hovione’s Strategy for Complexity, Speed, and Regional Supply Chains
3
FDA Grants Priority Review For Pfizer’s Marstacimab for Hemophilia A or B
4
PharmTech Weekly Roundup - February 6, 2026
5