

GMP agents report on old products, aseptic violations, and unexpected emotions.
Quality Systems
Latest News

Advertisement
Advertisement
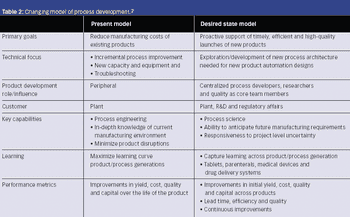
Operational excellence awaits, but only if you can implement PAT successfully.

Steps companies can take to help safeguard patients and the pharma supply chain.
Bernard Siegel, Founder and Executive Director of the Genetics Policy Institute (GPI), explains why lifting stem cell funding restrictions by executive order is only the first step towards safeguarding stem cell research.
President Obama's plan for increasing drug comparative effectiveness research is moving forward. The 15 members of the Federal Coordinating Council for Comparative Effectiveness Research, created to manage the $1.1 billion allocation designated for the research in Obama's American Recovery and Reinvestment Act of 2009, were named last week, according to a Mar. 19 press release from the US Department of Health and Human Services
Also, Hospira to reduce workforce; WuXi AppTech makes senior appointments; more...
In a case decided on Mar. 20, 2009, the US Court of Appeals for the Federal Circuit invalidated a US Patent and Trademark Office (PTO) Final Rule that governed the number of applications that parties may file to seek continued examinations of patent applications.
A bipartisan bill that would establish a regulatory pathway for the approval of biosimilars was introduced into the US House of Representatives last week.
Instead of protecting the public from unsafe drugs and contaminated foods, the Food and Drug Administration is a "hazard to public health," stated President Barack Obama in announcing his choices to head the agency and new efforts to improve food safety. Margaret Hamburg will be FDA's new commissioner, and Joshua Sharfstein principal deputy commissioner for drugs and medical products.
The US Pharmacopeial Convention (USP) and the National Institute for the Control of Pharmaceutical Biological Products (NICPBP), China's agency for overseeing the quality of large- and small-molecule drugs, signed a memorandum of understanding (MOU) to bolster the quality of medicines in China and in the countries that buy Chinese drug products, including the United States.
Ending a long, closely watched debate over the issue of federal preemption, the US Supreme Court on March 4, to uphold a $6.8 million Vermont Supreme Court decision of Diana Levine against Wyeth Pharmaceuticals (Madison, NJ).
The Synthetic Organic Chemical Manufacturers Association (SOCMA) said last week that Congress is likely to the include inherently safe technology (IST) measures in proposed chemical site-security legislation that is likely to be introduced in late winter or early spring.
Senators Byron Dorgan (D-ND), Olympia Snow (R-ME), John McCain (R-AZ), and Debbit Stabenow (D-MI) introduced a bill March 4 that would allow pharmacists and wholesalers to import prescription drugs from Australia, New Zealand, Japan, Switzerland and the European Union.
President Obama's Fiscal Year 2010 (FY 2010) budget includes support for a regulatory pathway for follow-on biologics.
On March 3, the US Food and Drug Administration released a draft guidance for Industry entitled "Clinical Pharmacology Section of Labeling for Human Prescription Drug and Biological Products: Content and Format."
In a press release dated Feb. 25, 2009, the US Food and Drug Administration charged that Ranbaxy Laboratories's (Gurgaon, Haryana, India) Paonta Sahib facility falsified data and test results in approved and pending drug applications.
FDA issued a notice in the Federal Register last week requesting comments on the automated collection of four additional pieces of information that are not available in the US Customs and Border Protection's (CBP) data set.

Broader disclosure of drug prices and conflicts of interest are central healthcare reform issues.

Industry has changed, but its basic tenets have not. INTERPHEX's RJ Palermo discusses a 7-step process to keep pharma moving forward.
The source of a problem reveals itself after some investigation, or it may crash down on you.
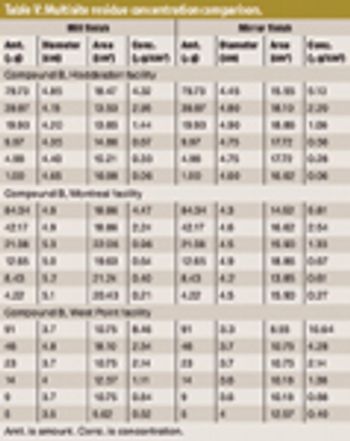
The author tests the ruggedness of VRL viewing conditions and defines optimal viewing conditions.

An updated book provides essential information for scientists who monitor microbial quality.

With the economy down and multinational firms capitalizing on patents in developing countries, India's R&D sector still has a long way to grow.
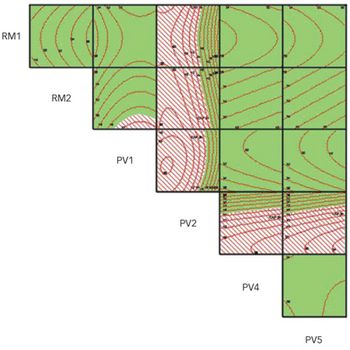
When applied as part of a structured approach, predictive modelling can provide deep process and product understanding, and can enable true, continuous process validation as envisioned by ICH guidelines.
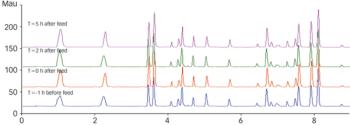
PAT guidance has been available from FDA for more than 4 years, but there have been no apparent breakthroughs in large-scale upstream production. Will companies consider using on?line chromatography to change this?
Advertisement
Advertisement
Trending on Pharmaceutical Technology
1
Drug Digest: Outsourcing Partnerships Fuel Global Biopharma Discovery and Scale-Up
2
Women in STEM: Early Phase Drug Development
3
FDA Grants Priority Review For Pfizer’s Marstacimab for Hemophilia A or B
4
PharmTech Weekly Roundup - February 6, 2026
5